长生链 基因大数据平台 IMC Genetic Data Cloud Platform
存储海量数据标本,通过分析为疾病的早期预防、早期筛查、早期诊断和药物研发提供重要依据,并为精准医疗奠定基础。

存储海量数据标本,通过分析为疾病的早期预防、早期筛查、早期诊断和药物研发提供重要依据,并为精准医疗奠定基础。
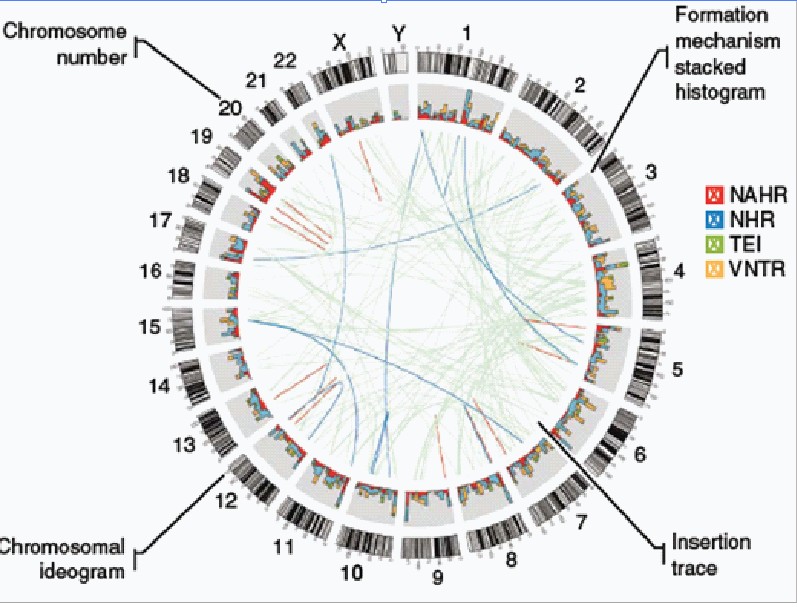
分析识别SV 的断点联结点(Breakpoint)
将Breakpoint按照可能形成的方式可以分类为以下几类:
(a)非等位基因同源重组型(non-allelic homologous recombination-NAHR);
(b)非同源重组(nonhomologous recombination-NHR),包括nonhomologous end-joining (NHEJ)和fork stalling /template
switching(FoSTeS/MMBIR);
(c)可变串联重复(VNTR)
(d)转座插入元件(TEI)。

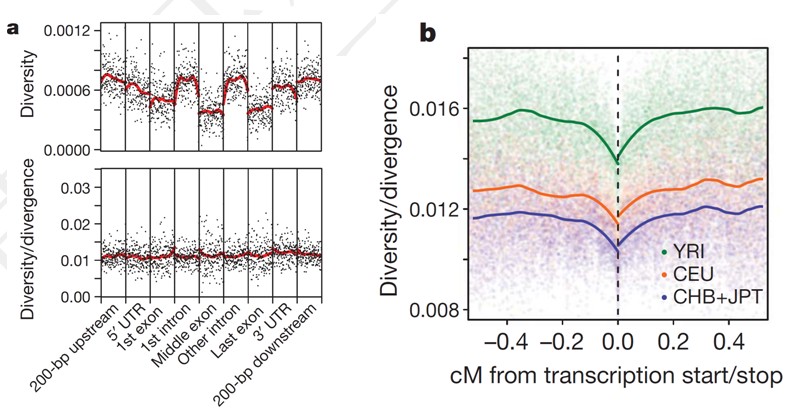
分析基因结构序列上不同区域的多样性(Diversity)与分歧进化(divergence)
根据基因型分析结果计算基因结构序列上的多样性程度,即杂合度(heterozygosity); 杂合度指标可以说明选择效应的存在以及局部变异的结构分布特征模式。我们将考虑基因5’UTR上游200bp
,5’UTR ,第一个外显子,第一个内含子,中间外显子,中间内含子,最末外显子和内含子,以及3’UTR及其下游200bp区域左右考察的范围(参见下图a)。
分析编码转录本的起始/终止位置临近区域的多样性和进化分歧度(参见下图b)。
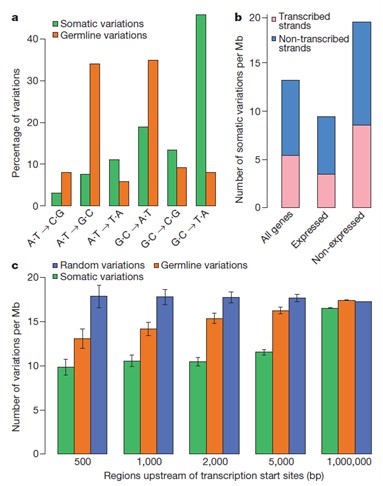
单核苷酸突变趋势与模式分析
分别统计在体细胞和生殖系细胞水平上的transversion的主要形式与各自所占比重(a);如果有表达谱数据,可以分析表达基因与非表达基因所分别具有的突变重排数目或者种类(b);转录起始位点上游区域的体细胞变异,生殖系germline变异以及随机变异的各自数目统计(c)和已知210种的不同肿瘤疾病的突变谱进行比较.

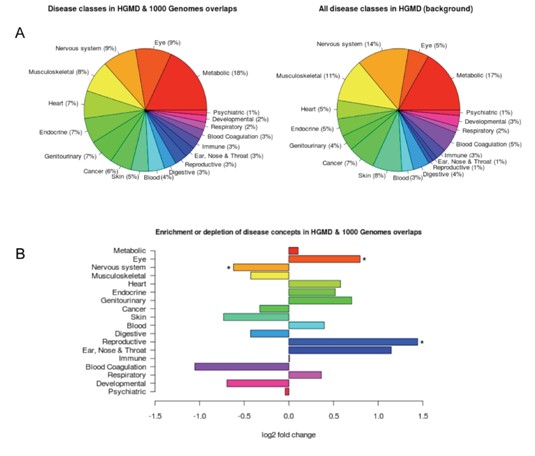
疾病变异体探测
将样本测序中分析得到SV与HGMD疾病变异体数据进行比对,得到交叉记录的错义和无义的SNP;通过将HGMD疾病关联突变与CUI(疾病概念分类标识数据库)比对获得HGMD中所有SV的疾病表型,并获得HGMD与测序数据分析得到的SV的疾病表型;并通过Fisher检验和Bonferroni多重假设检验校正计算样本SV所富集的疾病表型。
自然选择分析
我们通过测序所观测到的体细胞突变可能是经历了复杂的过程所成的。因此,我们在研究这些突变的起源,突变如何受到DNA修复机制的影响,以及在疾病发展与进化过程中突变的规律方面需要做深入的分析。自然选择一般在两个方面发挥作用,即保留有利于疾病发展进化的突变的同时限制其在基因组中重要功能区域发生突变,例如转录调控区域和编码蛋白质的区域。因此,(1)如果实验设计是将primary
disease与normal
control做比较的话,系统的分析可以解析复杂疾病在形成突变过程中可能的机制和自然选择的因素。(2)如果实验设计是基于病灶及其转移位置或者邻接位置样本作测序,我们可以构建突变进化与转移的模型解析突变的动态模式和基因组中不稳定态变异的模式。
正向选择的判定: 分析SNP,SNV区域的正向选择趋势,在进化和群体遗传水平解释SNV,SNP的功能性;对待control与case 组样本,我们分别采用不同统计算法计算SNP,CNV在各自样本中的差异,进而从中发现具有正向选择特征的SV。